Key Terms And Concepts Of Computer Validation
There are a number of terms and concepts that we need to get right before we start the detailed journey into validation of your software in the following articles in this series.
There are a number of terms and concepts that we need to get right before we start the detailed journey into validation of your software in the following articles in this series.
What is Validation?
Process validation is defined as:
Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specification and quality attributes [FDA Process Validation Guidelines, 1987].
This definition was modified by the PDA for a computerised system:
Establishing documented evidence which provides a high degree of assurance that a specific computer-related system will consistently produce a product meeting its predetermined specifications [PDA Technical Report 18, Validation of Computer-Related Systems, 1995].
The key concepts in the last definition above are:
- Documented evidence;
- High degree of assurance;
- Predetermined specification
There are other regulatory or quality guidelines from the European Union and the Organisation for Economic Development. Each regulation may have slightly different requirements but all come down to the same series of requirements: in general, validation is concerned with generating the evidence to demonstrate that the system is fit for the purpose you use it for and continues to be so when it is operational and that there is sufficient evidence of management control. This usually means that an action must be documented. Another feature of validation is to produce an auditable system; having the appropriate documentation to aid any audit or inspection.
The problem is how to respond to the requirement for computer validation. Any response should be:
- Scientifically sound
- Structured
- Provide adequate compliance
- Reflect the way you use the application
This latter point is most important, there is no point validating a function of a system that is not used.
Equally important, is the fact that one organisation’s use of an application can be markedly different from another’s use of the same software.
Computer validation must provide confidence in the system first and foremost to management and the users, secondly to an internal quality audit and thirdly to an external inspector. Inspectors only audit an organisation on a periodic basis; all others work in the organisation and use its computerised systems daily. The users must have the confidence in a system above all others, otherwise your investment will be wasted.
What is a Computerised System?
Virtually all equipment used in laboratory’s, clinical R&D or in production environments are classified as computerised systems. The key components are shown in Figure 1 [PDA Technical Report 18]. It is important to realise early in your project that if you are validating a computerised system, you don’t just concentrate on the computer hardware and software, as validation encompasses more requirements.
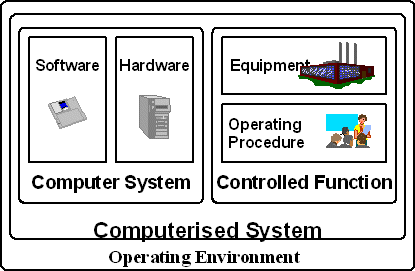
Figure 1: Elements of a Computerised System
The elements comprising a computerised system consist of a computer system and controlled function within the context of it operating environment.
The computer system consists of:
Hardware: The elements that comprise this part of a computerised system are the computer platform that the software application runs on such as:
- Workstation or server plus clients etc,
- Any network components such as hubs, routers, cables, switches and bridges. The system may run on a specific segment of a network or over a general segment of it, peripheral devices such as printers, plotters and connecting cables.
- Print servers
- Specialist peripherals such as bar code printers and readers etc
Software: This comprises several elements such as:
- Operating systems of the clients and server
- Network operating system.
- Application software and any utility software such as a database or reporting application such as Crystal Reports
The controlled function comprises:
- Equipment linked to the computerised system i.e. laboratory or clinical instrument or a production equipment. The interface can vary from a simple transmission of readings to complex control and data acquisition. Ideally the equipment connected to the data system should be qualified as part of the overall validation of the software, otherwise how do you know that you are generating quality results?
- Written Procedures: Trained staff should follow written SOPs as well as the manuals to operate the equipment and the data system software. This will include the computer department support staff if the application is a client server architecture.
Principles of Computer Validation
There are a number of principles that should be followed correctly during validation. The following are a summary of the main ones and these are intended to be practical issues that have arisen when validating computerised systems.
| System Owner is Responsible for Validation | The business owner of each system is responsible for the validation of that system. Whilst others may carry out validation on behalf of the system owner; the responsibility for validation cannot be delegated. |
| Risk Assessment | Does the system have to be validated is a key consideration at the start of any computerised system project. If the system is to be used to generate regulatory data, then validation is required, however if it is used for research purposes only then validation is not. Undertake a formal risk analysis and document the result. |
| Team Approach | Validation generally requires support from various functions and levels within the organisation e.g. scientists involved in using a system, system owner, quality assurance and if the system is networked, the IT department staff responsible for maintaining the server etc. All roles involved in validation must take responsibility for validation. |
| Validation Plan | There must be a formal and approved validation plan for each system. This needs to be written as early in the project as possible to avoid validation additional costs involved by writing documentation retrospectively that should have been written at the time the activity occurred. |
| Document Activities | All activities must be documented in reviewable forms. Today this documentation can be in either paper and / or electronic formats. Note carefully that it is not enough to observe the result of an activity or test, the politically correct term for this approach is informally documented; this leads to regulatory observations and warning letters. |
| Four Eyes Principle | All documents should be written and reviewed by at least two people (or two sets of eyes) to ensure that they are correct from both the technical and compliance perspectives. |
| Document Your Requirements | Your user or system requirements specification is your map through the system development life cycle and prevents you being seduced by technology or salespersons. Without this document, you cannot validate a computerised system. |
| Traceable Requirements | All functions and components of a system must be traceable to approved specification document(s). Furthermore, it must be demonstrated that these requirements are met within the implemented system. |
| Vendor Assessment / Audit to Assess Software Quality | Vendors must be assessed and if necessary audited. It is not adequate that another organisation has audited the vendor; this must be performed by your organisation, Furthermore, it cannot be assumed that products purchased from vendors are validated; all products (hardware, software, services) purchased must be checked for validation according to approved procedures. |
| Predefined Test Results and Acceptance Criteria | All testing must be based on comparison of actual results to expected results within defined and approved test scripts. Furthermore, acceptance criteria must be explicitly stated, not implied, and based on sound scientific principles. |
| Documented Operation | It must be demonstrated that operation of a system follows the system Standard Operating Procedures. These SOPs must be followed by the users and reflect current working practices with the system. |
| Independent Approval | The person approving key validation documentation must be independent of the validation team, the users and the developers of the system. A Quality Assurance involvement from the beginning of the project is essential. |
| Organised Archive | An archive for validation documentation must exist and it must be well organised. It must be possible to retrieve both physically and electronically archived documents accurately and quickly. This is essential to meet the requirements of 21 CFR 11 regulations. |
| Training and Ongoing Training | It must be demonstrated that all users, management, technical support and IT operations are trained in and are familiar with the system on an ongoing basis and applicable regulations. This will require initial and on-going training for all types of users (system manager, supervisor, user and IT support staff). |
| Standard Operating Procedures | The system must be operated using documented and approved Standard Operating Procedures. Further, and crucially, it must be possible to demonstrate that users continue to use the documented Standard Operating Procedures over time. |
| Change Management | Formal change management and configuration management procedures must be applied to all configuration items of the system e.g. hardware, application software, system software, training materials, SOPs and all documentation. |
| System Access Defined | Logical and physical access to the system, functions and the data must be clearly defined and validated. This needs to be updated regularly for compliance with 21 CFR 11 regulations if changes are made to access by users or functionality is increased. |
| Maintain Validation | Once validated, a system does not stay so automatically. The system owner needs to ensure that the system remains under control and changes need to be validated or revalidated when they occur, after the impact of the change has been assessed. Moreover, the system may need to be audited internally to ensure that the validation status has not changed. |
Assumptions and Misconceptions of Validation
Many people are familiar with validation in general terms and therefore a range of assumptions exist about it, but many these are incorrect or false. To help avoid these misconceptions, some of the more frequent ones are addressed in this section.
| We Bought a Validated System |
False! Any vendor product implemented in a particular environment becomes a unique item, as the combination of environment, parameters, configuration, data content, interfaces, user procedures etc. are unique. Note, as stated above, the system owner is responsible for validation and this cannot be delegated. The use of certificates of “validation” from vendors only apply to the portion of the system development life cycle that the vendor is responsible for. The system owner is responsible for the whole life cycle and, at best, these materials only provide a partial solution. |
| Partial Validation of the System | You cannot partially validate a computerised system, its is either all (validated) or nothing (unvalidated); see the FDA draft guidance document of General Principles of Software Validation for further information. |
| Long Term use Equals Validation | The fact that a system performs without problems for an extended length of time does not mean that the system is validated. To be validated a system requires documentary proof it meets predetermined validation criteria. See also comment 65 of the 21 CFR 11 preamble for further details. |
| Validation is a One-off Activity |
Validation is not a single event in a system’s life cycle as changes to the system inevitably occur, for example upgrade of application software or operating system. Therefore, on-going revalidation of a system is required until the system ceases operation. The data generated by the system need to be available for a batch’s shelf-life plus one year for GMP data or 15 years after the last launch in the last country for a system operating in R&D especially in the light of 21 CFR 11. |
| Validation Does not need Documentation | Oh yes it does! All activities contributing to validation of a system must be proven to have taken place i.e. documented either in paper or electronic means. If it’s not written, (approved and reviewed) the activity did not take place. |
| GMP = Giant Mass of Paper | The documentation needed to validate a system is little if any more than that required for good practice delivery of a computerised system not requiring validation. Further, references to vendor documentation can be used when these references include author, title, date/release number, and location. |
| Validation Equals Software Testing |
Wrong again! Firstly, a system includes more components than just software e.g. procedures, hardware, documentation and people. Secondly, other activities than testing are needed to prove a system functions as desired e.g. system specifications. Validation is a process that once started cannot be stopped. |
| Requirements are not Needed |
The definitions of validation above explicitly state that system requirements are required. In the absence of requirements:
|
| Just a Documentation Exercise | It is not adequate just to document validation features retrospectively. Validation must be proactively specified into a system. Further, it must be demonstrated that use of the system in practice continues to meet designed in validation features e.g. that Standard Operating Procedures are being followed. |
| Validation is a job for IT or QA/QC | Validation is the responsibility of the users of the system, in particular, the system owner who is legally responsible for validation. You can’t delegate this responsibility except due to incompetence. |
| Regulatory Bodies don’t care about IT Systems | Wrong yet again! There are increasing trends both to inspect IT systems and in the level of sophistication of such inspections. This is especially the situation with 21 CFR 11 regulations and the resulting warning letters. |
Computer Validation Roles and Responsibilities
There are three key roles in validating any computerised system, these are the users, quality assurance and the Information Technology (IT), where appropriate. Each role will be described below together with an outline of their responsibilities.
- Users: responsible for the overall validation of the system. This is achieved by defining the system's functions, selecting the system, verify its installation and to define and execute the validation plan. Users will need to have standard operating procedures written for operating and supporting the application, the user base must be trained and users must ensure that the complete documentation of the system is available for audit and inspection. Although the end user is responsible for these areas, they need help, advice and support in this. Active support by management is essential for making the resources available for the validation effort and to take the responsibility for authorising the use of the system in the regulated environment. Furthermore, management must encourage the participation of the Quality Assurance (QA) in this process.
- Quality Assurance: responsible for assistance in the interpretation of regulatory guidelines for computerised systems and how they apply to the spectrometer software. QA will review the key documentation produced during the validation effort. Monitoring of the testing and validation effort and offering assistance in developing SOPs, are additional roles and responsibilities for the QA. If there are any vendor audits to be undertaken, then the QA should be involved in the planning and execution of this activity. However, some QA personnel may not be very computer literate, but this must change as many regulations involving computerised systems require the active involvement of the QA.
- Information Technology: responsible for help in purchase, installation and operation of the spectrometer software for systems running on a network. If a group is not available or the users take on this role, then the responsibilities outlined here will be transferred to the users. Responsibilities will include running the hardware and software, backups, resolving problems etc. However, in offering support for a regulated spectrometer software, the IT group become bound by the regulations or guidelines that the laboratory work under. What is not often realised both by the users and the IS group is that any unauthorised change to the operating system or network will make a validated spectrometer software non-compliant.
External roles may also be involved these include the following:
- System Vendor: The system vendor should be able to help with advice on the sizing of the system, hardware needed for good performance, assistance with vendor audits and help with qualification of the system (Installation qualification and operational qualification only).
- Consultants: Consultants can offer advice on the overall validation process or specific portions of it. Use of consultants must be justified and managed to ensure value for money and benefit to the organisation.